Towards a Transferable Acceleration Method for Density Functional Theory
{kind=link}
Abstract
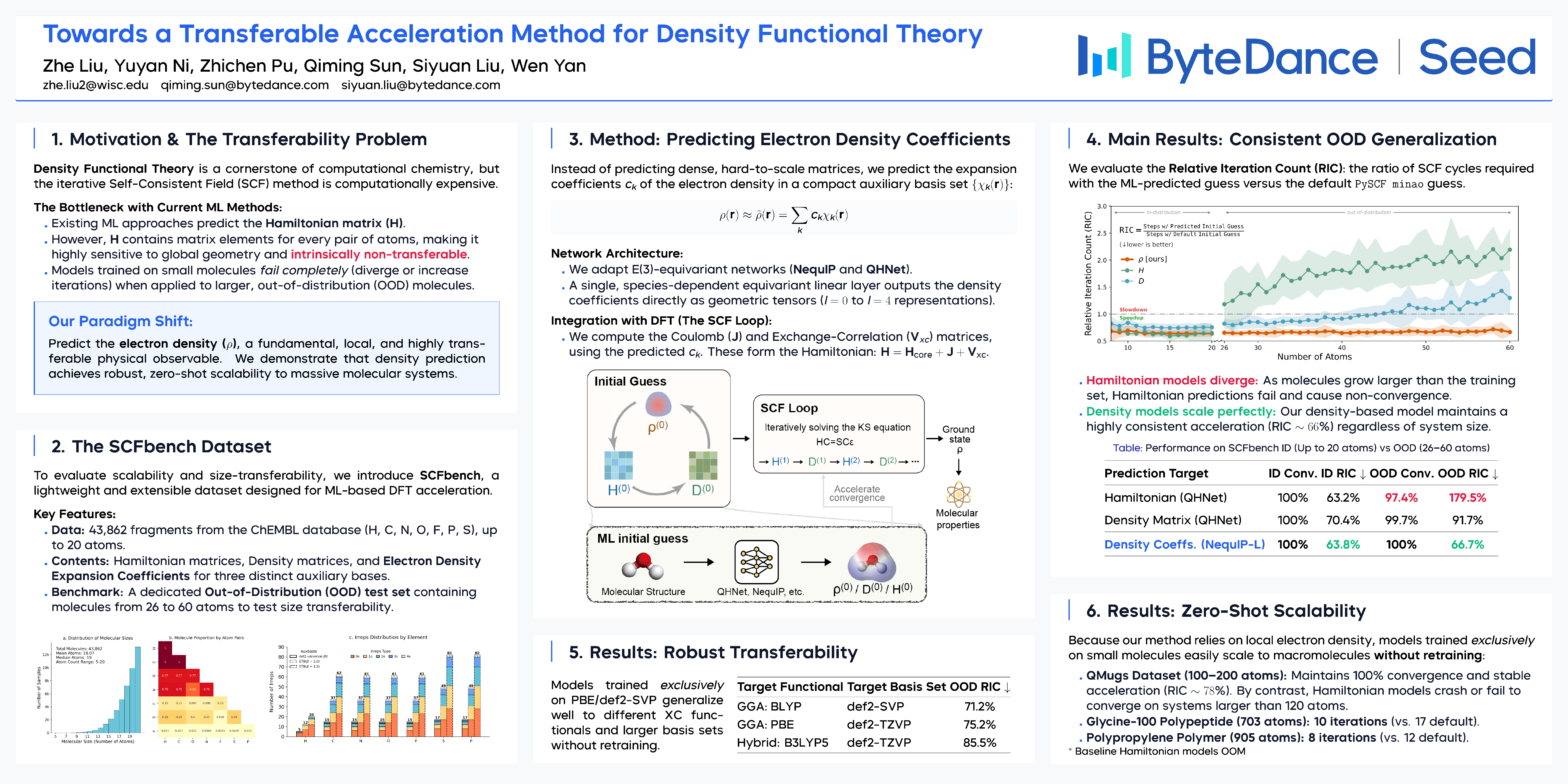
Recently, sophisticated deep learning-based approaches have been developed for generating efficient initial guesses to accelerate the convergence of density functional theory (DFT) calculations. While the actual initial guesses are often density matrices (DM), quantities that can convert into density matrices also qualify as alternative forms of initial guesses. Hence, existing works mostly rely on the prediction of the Hamiltonian matrix for obtaining high-quality initial guesses. However, the Hamiltonian matrix is both numerically difficult to predict and intrinsically non-transferable, hindering the application of such models in real scenarios. In light of this, we propose a method that constructs DFT initial guesses by predicting the electron density in a compact auxiliary basis representation using E(3)-equivariant neural networks. Trained exclusively on small molecules with up to 20 atoms, our model achieves an average 33.3% reduction in SCF iterations for molecules three times larger (up to 60 atoms). This result is particularly significant given that baseline Hamiltonian-based methods fail to generalize, often increasing the iteration count by over 80% or failing to converge entirely on these larger systems. Furthermore, we demonstrate that this acceleration is robustly scalable: the model successfully accelerates calculations for systems with up to 900 atoms (polymers and polypeptides) without retraining. To the best of our knowledge, this work represents the first and robust candidate for a universally transferable DFT acceleration method. We also released the SCFbench dataset and its accompanying code to facilitate future research in this promising direction.